Revisión clínica de 28 casos de incontinencia pigmentaria
Clinical review of 28 cases of pigmentary incontinence
Alina García García,I Iván Hernández García,II Norma de León Ojeda,III Marta Acosta Sabatés,IV Lurdes Marrón Portales V
IEspecialista
en Genética Clínica. Departamento de Genética Clínica. Hospital
Pediátrico Docente «William Soler». La Habana, Cuba.
IIEspecialista
en Genética Clínica. Departamento de Genética Clínica. Hospital
Pediátrico Docente «William Soler». La Habana, Cuba.
IIIEspecialista
de II Grado en Genética Clínica. Departamento de Genética Clínica.
Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
IVLicenciada
en Biología. Máster en Antropología Física. Departamento de Genética
Clínica. Hospital Pediátrico Docente «William Soler». La Habana,
Cuba.
VLicenciada en
Laboratorio Clínico. Departamento de Genética Clínica. Hospital
Pediátrico Docente «William Soler». La Habana, Cuba.
RESUMEN
INTRODUCCIÓN.
La incontinencia pigmentaria es una rara genodermatosis, presente
habitualmente en el sexo femenino, que se caracteriza por
alteraciones en los derivados del ectodermo superficial y del
neuroectodermo. El objetivo de esta investigación fue resumir las
características clínicas de esta entidad.
MÉTODOS.
Se revisaron 28 historias clínicas de niños atendidos en la consulta
de genética clínica del Hospital Pediátrico «William Soler» (Ciudad
de La Habana), que tenían diagnóstico clínico de incontinencia
pigmentaria. De las historias se tomaron los datos sobre el inicio y
evolución de la enfermedad, así como la información aportada por las
interconsultas de varias especialidades como dermatología,
estomatología y neurología. Cuando fue posible se hizo una
reevaluación clínica de los afectados.
RESULTADOS.
Solo 1 de los 28 pacientes era del sexo masculino. Entre las
primeras lesiones detectadas se encontraron 13 casos de vesículas
(43,3 %), 2 casos de eritema y 2 de descamación. Se encontraron máculas
de tipo hipercrómicas en 27 niños (96,6 %), hipocrómicas en solo 1
y verrugosas en 3. Las lesiones se distribuyeron en los miembros
inferiores en 22 casos (73,3 %) y en 19 casos en los miembros
superiores y en el tórax (63,3 %); en 21 pacientes (70 %) fueron
bilaterales. Con respecto a los anexos de la piel, se encontró
alopecia en 3 casos (10 %), hipodoncia en 8 casos (26,6 %) y
distrofia de las uñas en 3 (10 %). Se encontraron escleras azules en 6
casos (20 %) y estrabismo en 5 (16,6 %). Como expresión de daño del
sistema nervioso central se observó retraso mental en 12 casos (40 %)
y convulsiones en 6 (20 %).
CONCLUSIONES.
La incontinencia pigmentaria es una entidad heterogénea desde el
punto de vista clínico, pero es posible su reconocimiento por
alteraciones en la piel que atraviesan estadios previsibles. Es
necesario tener en cuenta que podría ser enmascarada por el color de
la piel o por el momento en que el paciente es examinado.
Palabras clave: Incontinencia pigmentaria, displasia ectodérmica, genodermatosis.
ABSTRACT
INTRODUCTION. The
pigmentary incontinence is an uncommon genodermatosis usually
present in female sex characterized by alterations in derivatives of
superficial ectoderm and of neuroectoderm. The aim of present
research was to summarize the clinical features of this entity.
METHODS.
Twenty eight medical records from children seen in the clinical
genetics consultation of "William Soler" Children Hospital (Ciudad de
La Habana) diagnosed with pigmentary incontinence. From the medical
records we got the data on onset and course of this disease, as well
as the information offered by inter-consultations of some specialties
including Dermatology, Stomatology and Neurology. When it was
possible a clinical re-assessment of involved was carried out.
RESULTS.
Only 1 of the 28 patients was of male sex. Among the first lesions
detected were the presence of vesicles in 13 cases (43,3%), erythema
in 2 cases and epidermis shedding in 2 cases. There were hyperchromic
maculae in 27 children (96,6%), hypochromic in only one and verrucous
type in three. Lesions were distributed in lower extremities in 22
cases (73,3%) in upper extremities and thorax in 19 cases (63,3%) and
in 21 patients (70%) were bilateral. Regarding the skin annexes
there was alopecia in 3 cases (10%), hypodontia in 8 cases (26,6%)
and nails dystrophy in 3 cases (10%), as well as blue sclera in 6
cases (20%) and strabismus in 5 cases (16,6%). As an expression of
damage in the central nervous system there was mental retardation in
12 cases (40%) and convulsions in 6 cases (20%).
CONCLUSIONS.
The pigmentary incontinence is a heterogeneous entity from the
clinical point of view but it is possible its recognition due to skin
alterations to go through foreseeable stages. It is necessary to take
into account that it could be masked by the skin color or by the
moment of physical examination of patient.
Key words: Pigmentary incontinence, ectodermal dysplasia genodermatosis.
INTRODUCCIÓN
La incontinencia pigmentaria (IP) es una rara genodermatosis que fue descrita por primera vez por Bloch y Sulzberger,1,2
y tiene una frecuencia de aproximadamente 1 cada 50 000 recién
nacidos. De herencia dominante ligada al cromosoma X, con usual
letalidad prenatal temprana en el varón afectado, provoca trastornos
en los derivados ectodérmicos, tanto del ectodermo superficial como
del neuroectodermo.3 Ocasionalmente se describen defectos
óseos, baja talla y trastornos en otros sistemas. El gen responsable
de la enfermedad se ha denominado NEMO (Nuclear Factor Kappa B
Essential Modifier [NF-κB essential modulator] o IKKγ [IκB
kinase-γ]) y se localiza en el locus Xq28.4,5 Cuando se
encuentra activo controla la expresión de múltiples genes, entre
ellos los que tienen que ver con la expresión de citoquinas y
chemoquinas que protegen a la célula contra la apoptosis.
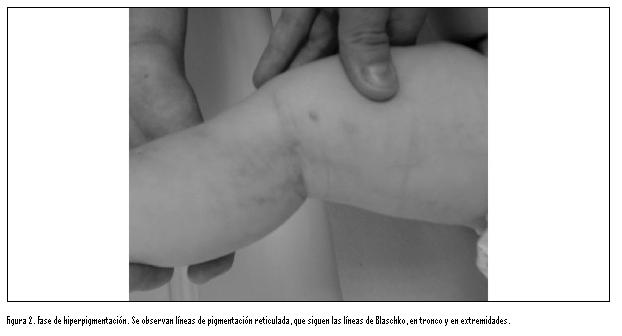
Las
lesiones epidérmicas clásicas de la enfermedad pueden pasar por 4
estadios: vesículas -generalmente perinatales-, lesiones verrugosas,
hiperpigmentación que sigue un patrón característico, y por último
lesiones hipocrómicas atróficas o cicatrizales (figura).
Este trabajo tiene
como objetivo resumir las manifestaciones clínicas de un grupo de
pacientes con diagnóstico clínico de IP.
MÉTODOS
Se
revisaron 28 historias clínicas de pacientes con diagnóstico clínico
de IP. La edad de los sujetos en el momento de su primera evaluación
osciló entre los 4 meses y los 5 años. Atendiendo al color de la
piel 12 sujetos eran blancos, 9 mestizos, 3 eran negros y en 4 no se
pudo conocer este dato.
De
las historias se tomaron los datos relevantes de inicio y evolución
de la enfermedad, así como lo aportado por las interconsultas de
varias especialidades como dermatología, estomatología, neurología,
entre otras. En los casos en que fue posible, se hizo una
reevaluación clínica de los afectados.
RESULTADOS
Del
total de sujetos de la muestra el 42,85 % eran blancos, el 32,14 %
mestizos y el 10,71 %, negros. En un porcentaje similar (14,30) el
color de la piel no fue descrito en la historia clínica.
En
cuanto a los antecedentes familiares se consideró importante la
existencia ya de la enfermedad en la familia, lo cual ocurrió solo en
4 pacientes (3 madres y 1 hermana afectadas) (14,28 %). Al analizar
la historia obstétrica de las madres se encontró que 10 de ellas
habían tenido abortos espontáneos del primer trimestre, antes o
después del propósito.
Las manifestaciones cutáneas (tabla 1)
de esta entidad son muy características y generalmente están
presentes en casi todos los pacientes, aunque no siempre son las
mismas. La forma de presentación más frecuentemente encontrada fueron
las vesículas (13-46,42 %). Aunque también se hallaron lesiones que
habitualmente no se describen en los estadios clásicos, como fueron
las lesiones hipercrómicas (27-96,42 %), que superaron ampliamente la
presentación de máculas verrugosas (3-10,71 %).
Tabla 1. Características de las lesiones cutáneas
|
Lesiones cutáneas
|
Número de casos
|
%
|
Primeras lesiones
| Vesículas
|
13
|
46,42
|
Eritema
|
2
|
7,14
|
Descamación
|
2
|
7,14
|
Máculas
| Hipercrómicas
|
27
|
96,42
|
Hipocrómicas
|
1
|
3,57
|
Verrugosas
|
3
|
10,71
|
Como se observa en la tabla 2
la distribución de las lesiones fue bastante heterogénea y en muchas
ocasiones los individuos mostraban uno o más sitios con lesiones,
aunque predominaron las manifestaciones cutáneas en los miembros
inferiores (22-78,57 %) y en los superiores y el torso (19-67,85 %).
En la mayoría de los casos (21-75 %) éstas fueron bilaterales. De las
distintas porciones del cuerpo la cara fue el sitio menos afectado
por las alteraciones en piel (2-7,14 %).
Tabla 2. Distribución de las lesiones cutáneas
|
Distribución
|
Casos
|
%
|
Cara
|
2
|
7,14
|
Cuello
|
5
|
17,85
|
Axilas
|
11
|
39,28
|
Tórax
|
19
|
67,85
|
Abdomen
|
16
|
57,14
|
Regiones inguinales
|
5
|
17,85
|
Miembros superiores
|
19
|
67,85
|
Miembros inferiores
|
22
|
78,57
|
Lateralidad de las lesiones
|
- Unilaterales
|
4
|
14,28
|
- Bilaterales
|
21
|
75
|
- No descrita
|
5
|
17,85
|
De los derivados del ectodermo superficial y anexos de la piel (tabla 3)
las más frecuentes fueron las anomalías dentales (18-59,9 %) y
dentro de este grupo la hipodoncia (8-26,6 %), que superaron a las
anomalías en los cabellos (8-26,6 %) y las uñas (3-10 %). Entre las
anomalías oculares las más llamativas fueron las escleras azules
(6-20 %) y el estrabismo (5-16,6 %). El retraso mental, indicativo de
repercusión de la entidad en el sistema nervioso central (SNC), fue
observado en 12 sujetos de la muestra (40 %) y el retardo del
lenguaje, relacionado con éste, se observó en 4 pacientes (13,3 %).
Otros defectos inespecíficos encontrados fueron la clinodactilia del
quinto dedo (2-6,6 %) y el dismorfismo facial (4-13,3 %).
Tabla 3. Afectación de otras estructuras
|
Órgano o sistema
|
Casos
|
%
|
Cabellos
| Alopecia(placas)
|
3
|
10
|
Pelo escaso
|
2
|
6,6
|
Otros defectos
|
3
|
10
|
Dientes
| Hipodoncia
|
8
|
26,6
|
Retardo en brote
|
4
|
13,3
|
Forma cónica
|
6
|
20
|
Uñas
| Distrofia
|
3
|
10
|
Ojos
| Escleras azules
|
6
|
20
|
Estrabismo
|
5
|
16,6
|
Microcórnea
|
3
|
10
|
Queratitis
|
1
|
3,3
|
Otros defectos
|
4
|
13,3
|
Sistema nervioso central
| Retraso mental
|
12
|
40
|
Convulsiones
|
6
|
20
|
Espasticidad
|
2
|
6,6
|
Retardo del lenguaje
|
4
|
13,3
|
Microcefalia
|
1
|
3,3
|
Macrocefalia
|
2
|
6,6
|
Hipoacusia
|
1
|
3,3
|
Óseas
| Sindactilia
|
3
|
10
|
Clinodactilia
|
2
|
6,6
|
Camptodactilia
|
1
|
3,3
|
Braquidactilia
|
1
|
3,3
|
Ectrodactilia
|
1
|
3,3
|
Dismorfia facial
|
4
|
13,3
|
Otros defectos
|
6
|
20
|
DISCUSIÓN
El
predominio de individuos de piel blanca en la muestra recopilada
podría estar dado por la coloración que adquieren las lesiones, que
podrían pasar inadvertidas en las pieles más oscuras. Llama la
atención que 10 de las madres tuvieron abortos espontáneos, por lo
que se hace necesario un exhaustivo examen físico a estas mujeres por
la posibilidad de estar afectadas de forma muy leve y de que estos
abortos correspondieran a fetos varones. Otra posibilidad de explicar
los abortos repetidos sería la presencia de un mosaicismo germinal no
detectado, el cual solo se confirmaría con la aparición de un nuevo
caso entre la descendencia de estas mujeres.
La
presencia de un varón afectado en la muestra podría explicarse por
varios fenómenos, entre ellos la presencia de un cromosoma X extra,
de alelos hipomorfos, un mosaicismo somático o tratarse de una
displasia ectodérmica anhidrótica con inmunodeficiencia.4 Esto último fue descartado.
Como
se puede apreciar, solo 13 de los 28 casos (46,42 %) comenzaron con
las típicas vesículas, por lo que la ausencia de este signo no
excluye la enfermedad cuando están presentes otras características.
En 2 casos se observó eritema y en 2 descamación; este último signo
no se describe en la bibliografía consultada. Chen observó lesiones
vesiculobulosas en el 90 % de los casos y verrugosas en el 70 % de
los pacientes entre las 2 y 6 semanas de nacidos.6 En un
metaanálisis realizado por autores canadienses la forma más común de
presentación hallada en el momento del diagnóstico también fueron las
lesiones vesiculobulosas (80 %).7 Los estadios 2
(lesiones verrugosas) y 3 (hiperpigmentación) se encontraron en el
72,5 % y 75 % de los casos respectivamente.7 Solo en el 15 % de los casos documentaron el estadio 4 (lesiones atróficas).7
Las lesiones hiperpigmentadas se observaron en casi la totalidad de
los casos (27-96,6 %) de esta serie, de forma similar a lo encontrado
(98 %) en los casos descritos por Chen.6 La atrofia de
las lesiones generalmente se encuentra en la adolescencia o en el
adulto joven. El último autor referido encontró atrofia de las
lesiones en el 42 % de los casos.6 En la muestra estudiada
no se encontró este estadio, lo que puede deberse a que estaba
compuesta, esencialmente, por lactantes y escolares.
En
la muestra estudiada predominaron las lesiones en los miembros
inferiores (22-73,3 %) y en el tórax (19-63,3 %) y los miembros
superiores (19-63,3 %), seguido del abdomen (16-53,3 %). Chen
encontró lesiones en el torso y las extremidades (90 %)
fundamentalmente.6 Las lesiones de piel fueron bilaterales
en los casos valorados de esta serie; sin embargo, Ardelean y Pope
encontraron que hasta en el 50 % de los casos las lesiones fueron
unilaterales.7
De
los anexos de la piel se encontró que hasta un 26,6 % de los
pacientes tuvieron defectos del pelo, cifras inferiores a las
encontradas por Chen que registró un 50 % de anomalías del cabello,
con especial incidencia de la alopecia del vértice.6 Los autores canadienses encontraron alopecia en el 30 % de los casos.7
Según
Chen las anomalías en la dentición aparecieron en más del 80 % de
los casos, aunque no se detallan específicamente los defectos.6 Ardelean y Pope encontraron un 40 % de anomalías dentales.7
En una recopilación de casos de Corea del Sur se encontraron
anomalías dentales en el 72,7 % de los casos, especialmente
hipodoncia.8 En esta serie se encontró menor incidencia de
esa anomalía (59,9 %), aunque fue la hipodoncia igualmente lo más
constante. De cualquier manera el bajo porcentaje de anomalías de la
dentición hallado podría estar dado por la edad de los pacientes de
la muestra analizada.
Las
anomalías del SNC (13-43,3 %), que en ocasiones quedan relegadas por
las más visibles alteraciones de la piel y las oculares (15- 59,9
%), fueron frecuentes. Chen reportó hasta un 40 % de afectación del
SNC.6 Ardelean y Pope documentaron alteraciones extracutáneas (manifestaciones del sistema nervioso
central y toma del ojo) en el 30 y 35 % de los casos respectivamente.7 Kim y cols. encontraron un 46,7 % de anomalías del SNC y 66,7 % de pacientes con defectos oculares.8 En general las anomalías más comunes halladas por ellos fueron la retinopatía y las convulsiones.8
Consideramos que en la muestra analizada no debiera dársele mayor
importancia al hallazgo de la esclerótica azul, si se tiene en cuenta
que es un derivado mesenquimatoso y que en su gran mayoría se debe a
inmadurez y es un signo frecuente en los lactantes sanos.
Otros
hallazgos (baja talla [3 pacientes], pie varo [1], foramen oval
permeable [1], hipotiroidismo congénito [1], retardo en la edad ósea
[1] y fístulas preauriculares [2]) no se recogieron en la tabla 2 y
no se encontraron en la literatura médica consultada.
La
IP es una entidad extraordinariamente heterogénea desde el punto de
vista clínico, pero su reconocimiento es posible por las alteraciones
presentes en la piel, las cuales atraviesan por estadios bastante
previsibles. Es necesario tener en cuenta que podría ser enmascarada
por el color de la piel del individuo o por el momento en que el
paciente es traído a consulta. Finalmente puede decirse que no existe
un solo derivado ectodérmico que no sea afectado por la alteración
en la expresión del gen NEMO.
REFERENCIAS BIBLIOGRÁFICAS
1.
Bloch B. Eigentumliche bischer nicht beschriebene pigmentaffektion
(incontinentia pigmenti). Schweiz Med Wochenschr 1926;7:404-5.
2.
Sulzberger MB. Uber eine bischer nicht beschriebene congenitale
pigmentanomalie (incontinentia pigmenti). Arch Dermatol Syph (Berl).
1928;1(54):19-32.
3. Pascual-Castroviejo I, Roche MC, Martínez Fernández V, Pérez Romero M, Escudero RM, García-Peña JJ, et al. Incontinentia pigmento: RM demostration of brain changes. Am J Neuroradiol (AJNR). 1994;15:1521-7.
4. Aradhya S, Courtois G, Rajkovic A, Levy M, Isral A, Nelson DL, et al.
Atypical forms of incontinentia pigmenti in male individuals result
from mutations of a cytosine tract in exon 10 of NEMO (IKKgamma). Am J
Hum Genet 2001;68:7657.
5. Aradhya S, Woffendin H, Jakins T, Espósito T, Smahi A, Shaw C, et al.
A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma)
gene accounts for the vast majority of incontinentia pigmenti
mutations. Hum Mol Genet 2001;10:217-9.
6.
Harold Chen. Incontinencia Pigmenti. In: Harold Chen, ed. Atlas of
Genetic Diagnosis and Counseling. New Jersey: Humana Press Inc.;
2006. Pp.539-44.
7.
Ardelean D, Pope E. Incontinentia pigmenti in boys: a series and
review of the literature. Pediatr Dermatol. 2006;23:523-7.
8. Kim BJ, Shin HS, Won CH, Lee JH, Kim KH, Kim MN, et al. Incontinentia pigmenti: clinical observation of 40 Korean cases. J Korean Med Sci. 2006;21:474-7.
Recibido: 27 de mayo de 2010.
Aprobado: 16 de agosto de 2010.
Alina García García. Hospital Pediátrico Docente «William Soler». San Francisco y Perla Altahabana. Ciudad de La Habana, Cuba.







{kind=link}